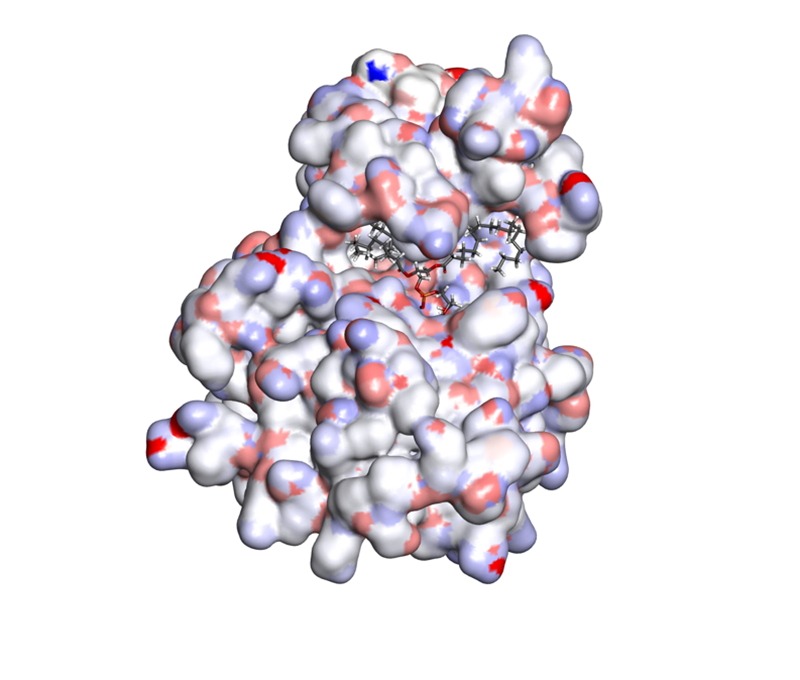
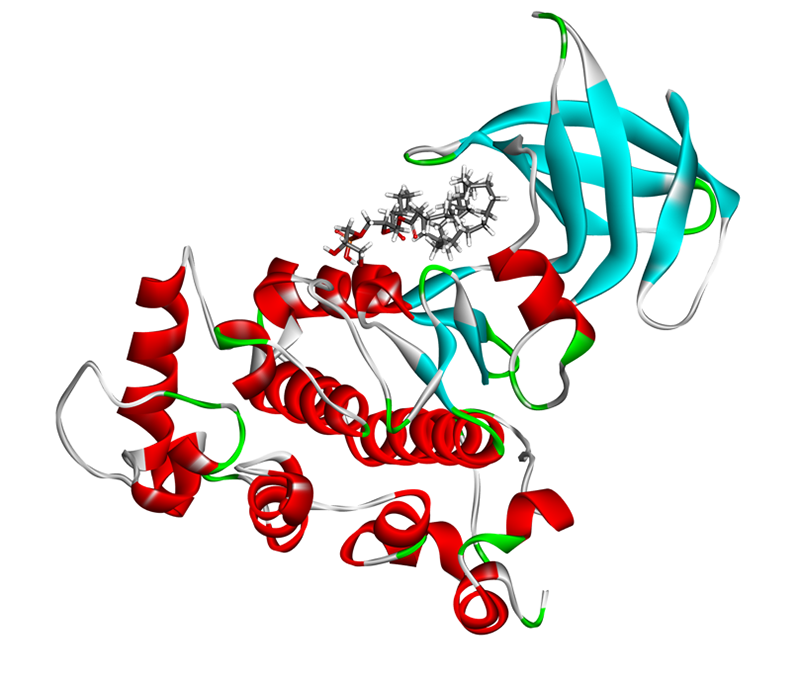
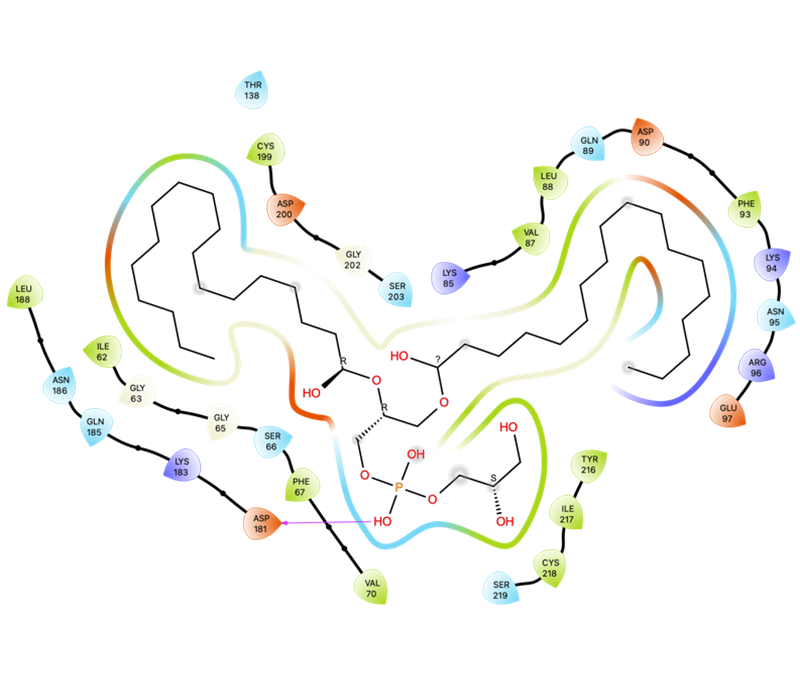
Molecular docking is used to predict the interaction between small molecules and biological macromolecules (drug molecule-protein) or between biological macromolecules (protein-protein or DNA/RNA- protein). Based on the principles of geometric matching, charge interaction and hydrophobic interaction between molecules, this technique simulates the spatial fitting process of two molecules to evaluate their binding tendency and stability. In drug design, molecular docking is used to predict the binding affinity between drug molecules and target proteins, so as to guide drug design and optimization. The application of this technology is not limited to drug development, but also includes understanding the catalytic mechanism of enzymes and protein-protein interactions. It is an important tool in the field of biochemistry and molecular biology.