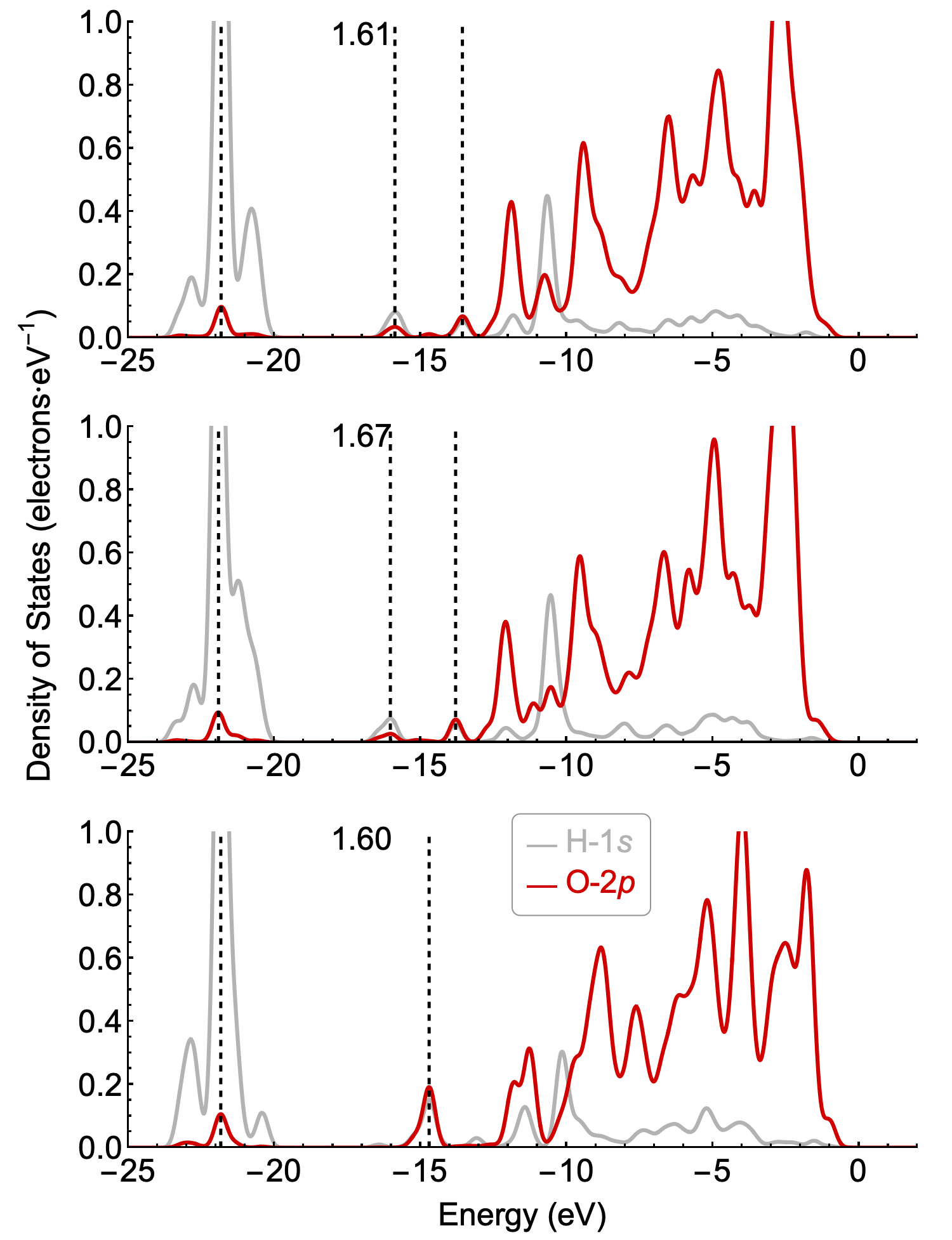
First principle (First Principles) here refers to its application in simulation methods. Its theoretical basis is deeply rooted in quantum mechanics and is mainly used to analyze and predict electronic behavior. This method is also known as ab initio algorithm (ab initio), and its core feature is that it does not depend on any empirical parameters. It only calculates the various properties of the material system by a given atomic number Z, combined with the basic physical constants-Planck constant h, speed of light c, electron charge e, Boltzmann constant kB and electron mass me--. The remarkable feature of the first principle is that it is independent of the experimental data and can accurately predict various properties in a certain scale. At present, this method has been widely used in physics, chemistry, life science, material science, catalysis, environmental science and other fields.