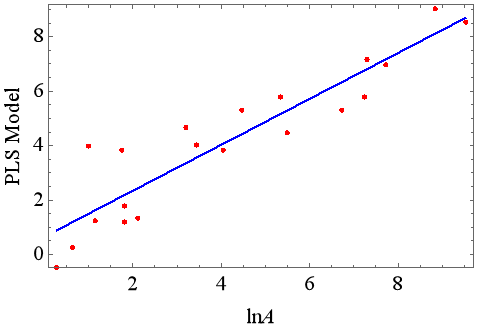
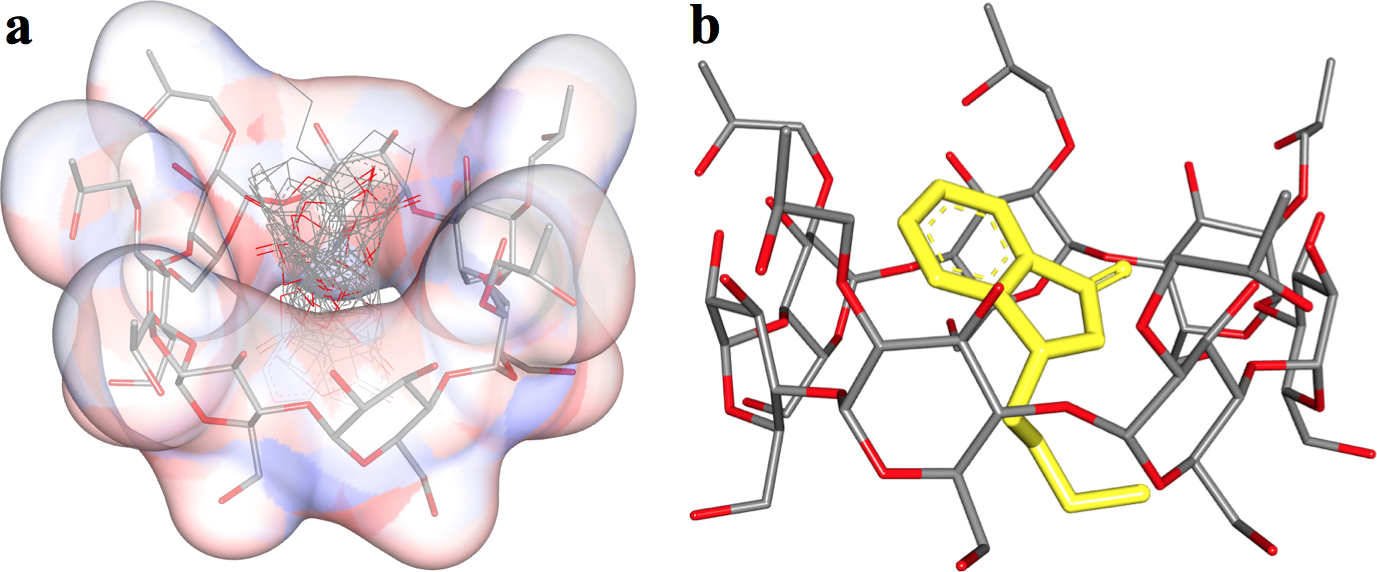
In the 3D-QSAR modeling process, we investigated the three-dimensional structural characteristics of 20 drug molecules, including ALogP, molecular weight, number of rings (including aromatic rings), number of hydrogen bond donors and acceptors, number of flexible bonds, polarized surface area of molecular fragments, torsional energy, solvent accessible surface area, molecular volume, and other relevant molecular fingerprint information. These characteristics relate to the number of times a drug molecule is solubilized by a particular solvent. Our model successfully predicts the solubilization factor of a drug molecule in a specific solubilizer, and the error between the predicted results and experimental data and other simulated values is within 5%, demonstrating the accuracy and reliability of the model.