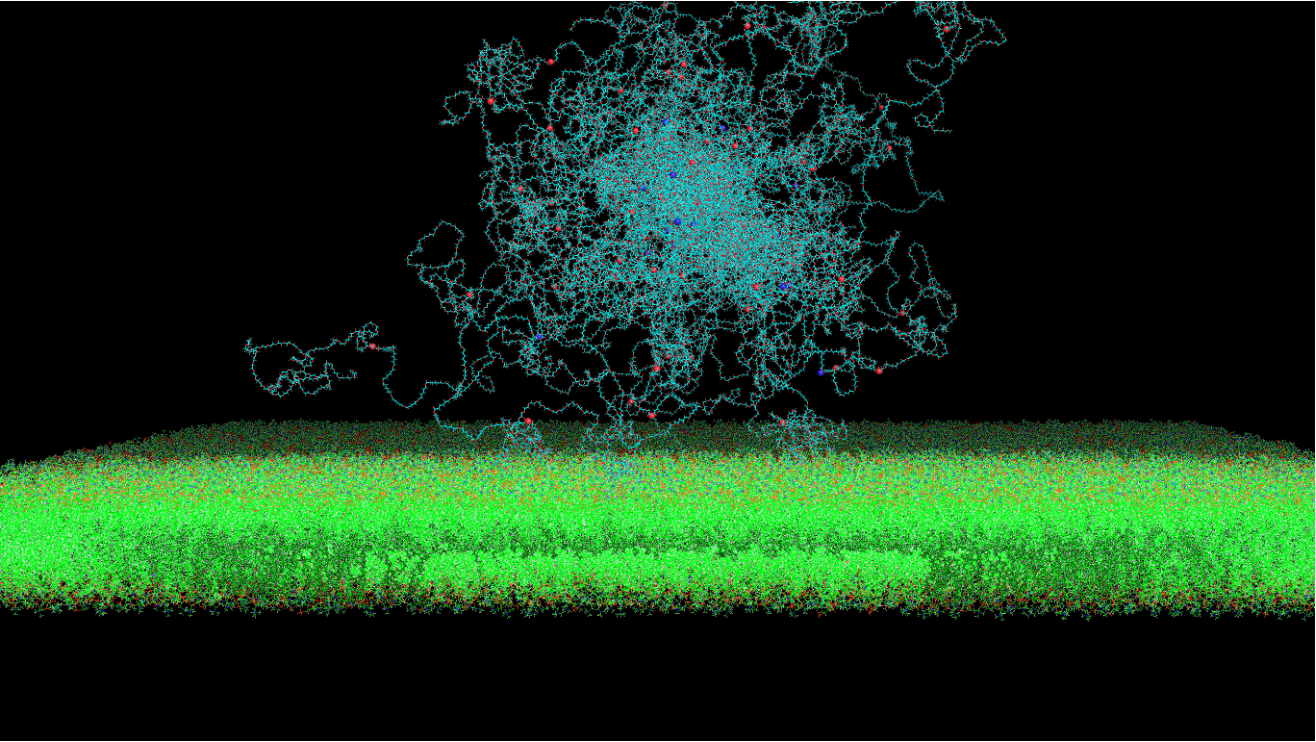
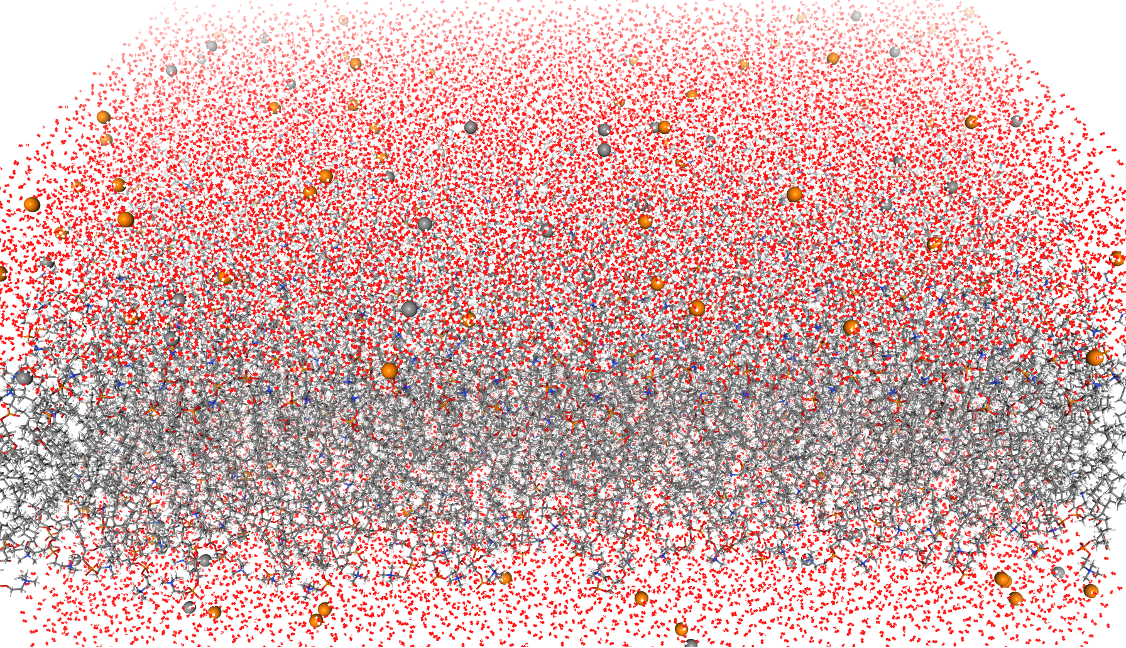
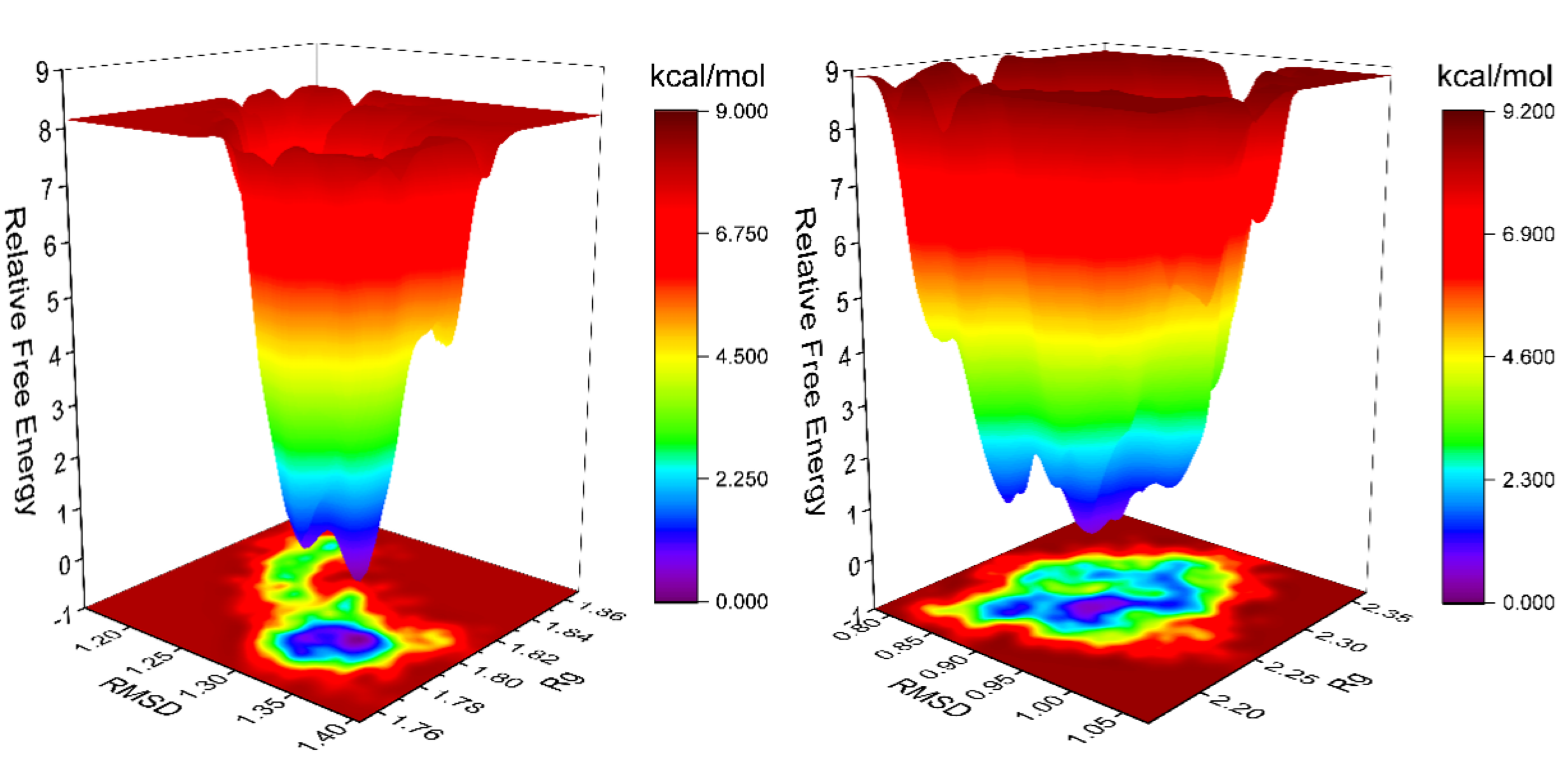
分子动力学(Molecular Dynamics, MD)在这里特指基于牛顿力学的模拟方法。与基于量子力学的第一性原理方法不同,MD模拟通过构建力场和求解能量函数来实现,从而在模拟中忽略了电子细节的更深层次考量。这种方法的一个显著优势是,在相同的硬件条件下,能够处理比第一性原理方法更大规模的体系。通过MD模拟,研究者能够获得体系原子的运动轨迹,深入观察原子层面的微观动态。这种动态模拟使我们能够从分子水平理解生物大分子的运动与功能、蛋白质与小分子间的相互作用机制以及纳米材料的分子自组装过程等。作为实验方法的重要补充,分子动力学模拟已广泛应用于物理、化学、生物医药和材料科学等多个领域。